IGEM Notebook (6/2/09 - 7/31/09)
Contents
- 1 --Leland 09:28, 2 June 2009 (EDT)
- 2 --Leland 17:35, 3 June 2009 (EDT)
- 3 --Leland 10:13, 4 June 2009 (EDT)
- 4 --Leland 12:02, 5 June 2009 (EDT)
- 5 --Leland 12:35, 8 June 2009 (EDT)
- 6 --Leland 08:54, 9 June 2009 (EDT)
- 7 --Leland 11:57, 10 June 2009 (EDT)
- 8 --Leland 09:28, 11 June 2009 (EDT)
- 9 --Leland 12:27, 12 June 2009 (EDT)
- 10
--Leland 09:28, 2 June 2009 (EDT)
Today Alyndria and I worked on answering the question of how do we insert and construct the logical clauses (LCs) into the reporter gene. We outlined two processes for doing the LC inserts. One as a BioBrick, and the other only changing the first part of the gene. We feel the second one is much better because there are only two parts (of the gene) to be linked together, and only the first part has to be edited.
Tomorrow Todo List
1. Finalize and upload the document created today
2. Begin work on which tRNA suppressor we should use.
--Leland 17:35, 3 June 2009 (EDT)
I worked on putting together a document showing which tRNAs we should use. Will came up with an alternative method for how we will insert the logical clauses (or anything); however, we have yet to create a document showing this method. We decided we should order all the tRNAs except for the wobble one (11 total), so that we can begin testing them. I put together the ATG and 5mer sequences for preliminary tests on our system. These sequences will be used to test to see if our idea of having one suppressor tRNA bind to mRNA and alter the reading frame, allowing a protein to be expressed, will work. Davidson ATG+5mer BioBrick Sequences
Tomorrow Todo List
1. Finalize and upload the document on which tRNA suppressor we should use
2. Check the ATG+5mer sequences with the reporter genes with Shashank
3. Design the other surpressor tRNA encoding biobrick
4. Update the document created yesterday with the new plan
--Leland 10:13, 4 June 2009 (EDT)
Using Olivia's program, Shashank and I went through the genes RFP and Tet resistance looking for other 5mer sites (in the reading frame that would occur if no 5mer binds at the designed "LC" site). Thankfully, no unwanted 5mers show up in that reading frame. However, we discovered that in the scar in the reading frame if the 5mer does bind at the correct place, is TAG which is a stop codon (UAG). We spent the rest of the day working on this problem. I also updated Inserting 5mers.
Tomorrow Todo List
1. Choose a reporter gene... needs to have a restriction site early on
2. Work out ideas to overcome TAG (UAG) in reading frame
3. Finish Question
4. Finalize and upload the document on which tRNA suppressor we should use
--Leland 12:02, 5 June 2009 (EDT)
I fleshed out CR/Restriction enzyme solution to the TAG codon. This method is similar to the 2nd process described in the Inserting 5mers document. The only difference is the method in which the front segments are generated is PCR. Then Romina and I obtained the registry numbers for the cells we will need to begin culturing Sunday afternoon for Monday in the lab.
Promoter
pLacIQ1 - K091112
968: 4-4, 49
969: 4-4, 50
pLac1Q - K091111
477: 3-4, 22
813: 1-4, 28
pLac + RBS – S03511
440: 3-2, 7
RBS
RBS (strong) – B0030KH
874: 4-4, 14
RBS – B0034
421: 2-1, 1
422: 2-1,
449: 2-1, 13
533: 1-3, 97
1056: 2-5, 57
Reporter Gene
RFP - E1010
441: 2-1, 8
445: 2-1, 10
TetR – C0040
505: 3-4, 44
512: 3-4, 51
***Note 6/8/09 this TetR part was incorrect. We needed Tet resistance not the Tet repressor gene.
Optional things we may want
pLsrR - K091114
812: 1-4, 27
pLac+RBS+RFP – I715039
426: 3-1, 1
429: 3-1, 2
Tomorrow Todo List
1. Design Primers
2. Do miniprep
3. Finalize and upload the document on which tRNA suppressor we should use
--Leland 12:35, 8 June 2009 (EDT)
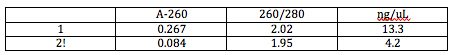
We finalized and ordered the primers and the tRNA suppressor gene oligos needed for our fist set of experiments Davidson Primers and Oligos. We then did a mini prep to prepare the bioparts we need (listed on previous post). The results for the ones I prepared are as shown.
K091111 - 1 part solution/ 3 part dH2O
Absorption @ 260 - 0.843
260:280 - 2.00
ng/µl - 42.1
S035710 - 1 part solution/ 3 part dH2O
Absorption @ 260 - 0.817
260:280 - 1.98
ng/µl - 40.9
Tomorrow Todo List
1. Finish with the cells in the tube
2. Finalize and upload the document on which tRNA suppressor we should use
--Leland 08:54, 9 June 2009 (EDT)
Notes on the processes
Plasmid Digestion 3.Buffer (Always 10%) Determined by which restriction enzymes are used. Check chart for BioBricks enzymes. Buffer 2.DNA 4.Restriction Enzymes When pipetting, barely touch top of liquid with pipette (it will grab on to outside) Must keep cold... very sensitive to temp changes. Must be less than or equal to 10% of the final volume Are most sensitive, so add last 1.dH2O ***If you have to make alot (maybe 10) make a cocktail of everything but DNA. In this cocktail do one extra. (so if you had 10, make enough for 11) Add cocktail to tubes with DNA. Example: Buffer (Always 10%) - 2 µL DNA - 3 µL Restriction Enzyme 1 - 1 µL Restriction Enzyme 2 - 1 µL dH2O - 13 µL FINAL VOLUME - 20 µL Usually use 20 µL
Gel Electrophoresis Which gel to use?... Find base pairs and use website Gel Concentration Site Memorize 1% solution = 1 g/100 mL Need to know: total volume of cast (60 mL for Campbell lab) Example: Need 0.8% solution (1 g/100 mL)x 0.8 = 0.8 g/100 mL (0.8 g/100 mL)x (0.6/0.6) = 0.48 g agrose/60 mL buffer
Data From Today
Plasmid Digestion Figures and Respective Gels

We prepared two gels. The first was 0.8% and the second was 2.5%. These two percentage of gels are close enough to the actual percentages so that we can fit all of the samples on these two. The plasmids of the BioBrock parts were digested with EcoR1 and Pst1.
2.5%

0.8%

The parts in each row are given in this table:
![]()
A breakdown of the molecular weight marker can be found here
We then cut the respective plasmids we need to purify out of the gel. These plasmids were the I715039, S03710, I715039, and J31007. It is important to note that all of these plasmids are the same because the biobricks were cut out of them with EcoR1 and Pst1
Additionally, we placed the cells containing the plasmids of the parts we purified on a agar plate so that they will survive. The plate was labeled as "LBAMP-AMC-9 June" and numbered as follows.
{kind=link}
1. B0030 AMC 2. K091111 3. S03710 4. I715039 OEH 5. S03511 ORH 6. I715039 AST 7. B0030 SDP 8. K091112 9. J31007 LJH 10. J31007 REC 11. S03511
Additionally Romina and I worked on figuring out what is wrong about the LacIQ promoter and the Lac IQ1 promoter. The top two runs on the 2.5% gel according to the registry should be even. However, they are obviously not. After looking at Pallavi Penumetcha paper on these promoters, we believe the parts listed in the registry are incorrect. We have emailed her and are working on solving this problem.
Tomorrow Todo List
1. Finish purifying the plasmids
2. Search for proteins that snip out amino acids before the final secondary structure forms
3. Finalize and upload the document on which tRNA suppressor we should use
--Leland 11:57, 10 June 2009 (EDT)
We ran a gel again for the S03511 because our previous gel seemed incorrect; however, we obtained similar results. The gel was the optimal concentration for the size of the DNA at 1.8%, and we obtained similar results to our first gel. We concluded the DNA part sample was bad and removed it from the catalog.

We then purified the 4 plasmids with the biobricks cut out of them, taken from the gel yesterday. We mixed all four plasmids in one tube. The following is the data from the nanodrop test and information on the samples.
Samples - Weight(g) - Buffer (3x weight:µL) 1 - 0.12 - 360 2 - 0.10 - 300 3 - 0.21 - 620 4 - 0.23 - 690
NanoDrop Absorption @ 260 - 0.172 260/280 - 2.28 (higher than expected) ng/µL - 8.6 (usually lower for gel purification)
We then created and prepared AMP agar
We also located parts we need in previous mini-preps. These parts are
pLac + RBS - J04500 pBad - I13453 pBad + RBS - J3102
Tomorrow Todo List
1. PCR and oligos assembly when the parts arrive
2. Prepare the rest of the plasmid inserts for the LCs so we only have to add the enzymes BAMH1 and Noc1
3. Search for proteins that snip out amino acids before the final secondary structure forms
4. Finalize and upload the document on which tRNA suppressor we should use
--Leland 09:28, 11 June 2009 (EDT)
We determined the buffer and the amounts of substances needed to make the holes in the reporter gene plasmid that we will place the LCs into. Because we are using alot of DNA, we increased the final volume to 40 µL instead of 20 µL.
Concentration Calculations:
310.4 ng/1 µL = 2500ng/x µL
x = 8.05 µL
This table show compatible buffers. We will use E buffer for BAMH1 and H buffer for Nco1


We transformed the cells with the parts we located yesterday in previous years mini-preps.
We ran PCR with the primers we received today to generate the LC inserts for RFP and Tet, shown in this table.

I was responsible the RFP and Tet CCCUC LCs. Olivia and Shamita diluted part of the RFP and Tet genes for template DNA by 1/100. The original concentration of the template DNA was determined yesterday by the nanodrop. Here are my calculations.
RFP
(310.4 ng/µL)(1/100)=3.104 ng/µL
(3.104 ng)x=(ng)µL
x=0.3221 µL RFP
Tet
(163.6 ng/µL)(1/100)=3.104 ng/µL
(1.636 ng)x=(ng)µL
x=0.611 µL RFP
Because the primers came in a 100 µM solution, we only have to add 1 µL
RFP CCCUC PCR dH2O = 47.6 µL Master Mix = 50 µL Temp DNA = 0.3 µL Forward Primer = 1 µL Reverse Primer = 1 µL Tet CCCUC PCR dH2O = 47.3 µL Master Mix = 50 µL Temp DNA = 0.3 µL Forward Primer = 1 µL Reverse Primer = 1 µL
We also preformed the oligo assembly for the tRNA suppressor gene. Again, we split them up and I was in charge of the CCCUC oligos.
Oligo parts = 1 µL (so x7) Buffer = 2 µL dH2O = 11 µL Total volume = 20 µL
Additionally we digested a RFP (S0371) plasmid sample with EcoR1 and Nco1 and a Tet (I713039) plasmid sample with BamH1 and EcoR1.
- All of the processes today were done according to the lab procedures.
Tomorrow Todo List
1. Purify PCR parts
2. Insert modified RFP and Tet end
3. Search for proteins that snip out amino acids before the final secondary structure forms
4. Finalize and upload the document on which tRNA suppressor we should use
--Leland 12:27, 12 June 2009 (EDT)
I spent most of the morning on calculations on for the gel used to purify the reporter genes and as an initial verification of the PCR with the rest of the team. We then calculated the amount of insert we needed for ligations. We performed the ligations, transformed them, and stored the cells in the incubator. Lyn will take them out tomorrow.
Digested Reporter Gene 0.4% Gel (in order from top to bottom) I715039 Marker S03710
0.4% gel

PCR Gel 1.45% (in order from top to bottom) #1 - RFP_CUAGU #7 - RFP_CCACU #2 - Tet_CUAGU #8 - Tet_CCACU #3 - RFP_CCCUC #9 - RFP_CCAUC #4 - Tet_CCCUC #10 - Tet_CCAUC #5 - RFP_CGGUC Marker #6 - Tet_CGGUC
1.45%

Something must have gone wrong with Tet_CUAGU preparation, because all of the others worked. Shamita and Romina prepared DNA for another gel tomorrow.
Tomorrow Todo List
1. Verify, clean, digest, clean, and ligate PCR
2. tRNA's
3. Search for proteins that snip out amino acids before the final secondary structure forms
4. Finalize and upload the document on which tRNA suppressor we should use